Extract and estimate the best supported model from a phylogenetic path analysis.
Source:R/phylopath.R
best.RdExtract and estimate the best supported model from a phylogenetic path analysis.
best(phylopath, ...)Arguments
- phylopath
An object of class
phylopath.- ...
Arguments to pass to phylolm::phylolm and phylolm::phyloglm. Provide
boot = Kparameter to enable bootstrapping, whereKis the number of bootstrap replicates. If you specified other options in the original phylo_path call you don't need to specify them again.
Value
An object of class fitted_DAG.
Examples
candidates <- define_model_set(
A = NL ~ BM,
B = NL ~ LS,
.common = c(LS ~ BM, DD ~ NL)
)
p <- phylo_path(candidates, rhino, rhino_tree)
best_model <- best(p)
# Print the best model to see coefficients, se and ci:
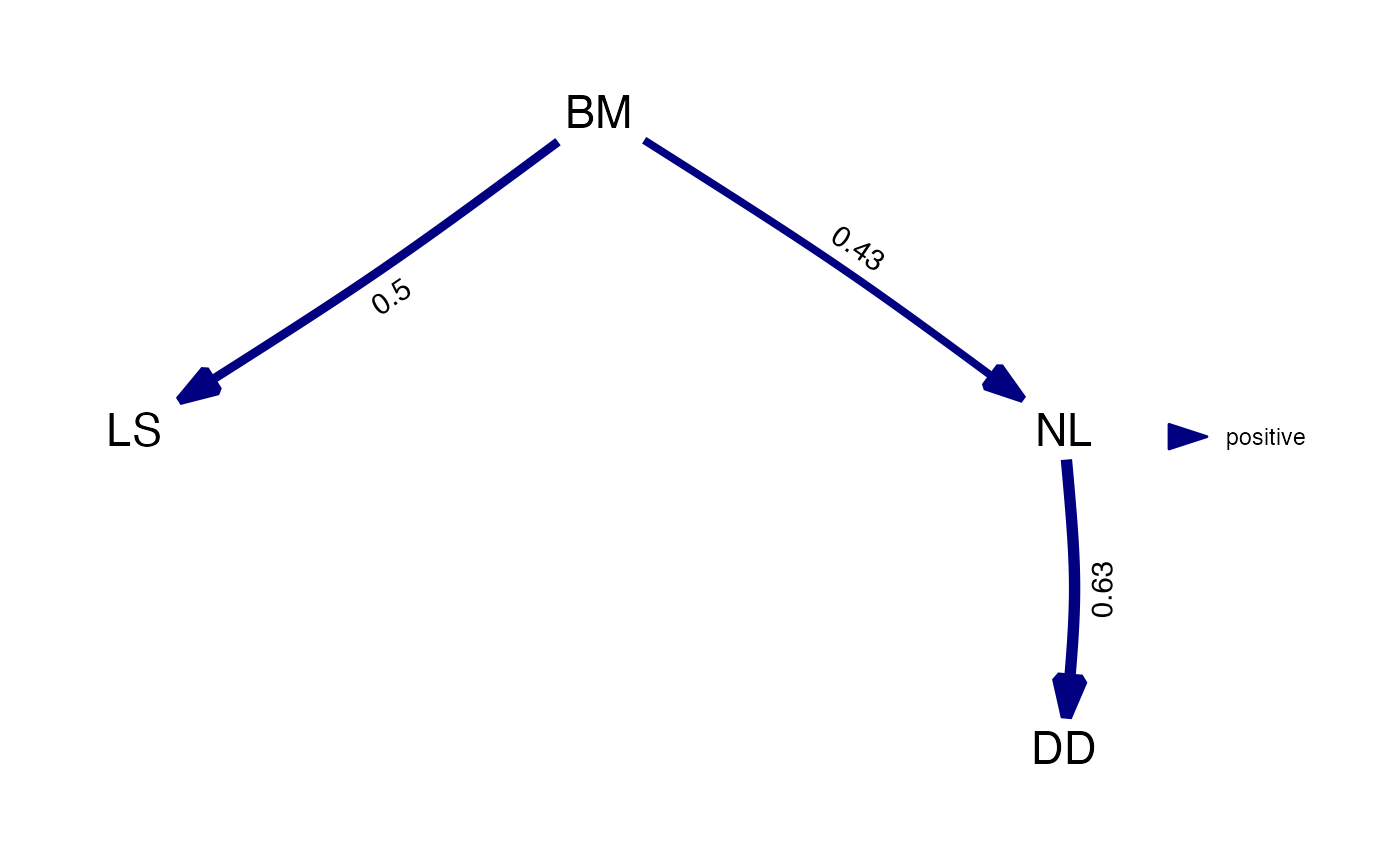
best_model
#> $coef
#> BM LS NL DD
#> BM 0 0.4973937 0.4303443 0.0000000
#> LS 0 0.0000000 0.0000000 0.0000000
#> NL 0 0.0000000 0.0000000 0.6285344
#> DD 0 0.0000000 0.0000000 0.0000000
#>
#> $se
#> BM LS NL DD
#> BM 0 0.08934185 0.08868688 0.00000000
#> LS 0 0.00000000 0.00000000 0.00000000
#> NL 0 0.00000000 0.00000000 0.08006703
#> DD 0 0.00000000 0.00000000 0.00000000
#>
#> attr(,"class")
#> [1] "fitted_DAG"
# Plot to show the weighted graph:
plot(best_model)